Vascular and pigmented neurocristopathies
Chef d'équipe
H. ETCHEVERS
H. ETCHEVERS
Des malformations présentes à la naissance sont par définition congénitales, Quand elles impliquent une population de cellules souches qui s'appelle la crête neurale, ces maladies congénitales relèvent des neurocristopathies. Des formes fréquentes touchent préférentiellement le palais, les yeux et/ou le coeur, mais il y a des douzaines d'autres maladies plus rares qui touchent les systèmes plus disseminés tels la peau ou le système nerveux périphérique. Alors qu'on connait depuis une dizaine d'années de plus en plus de bases génétiques des neurocristopathies, la plupart restent encore aujourd'hui sans diagnostic moléculaire et parfois sont mal définies en tant qu'entités cliniques.
Nous avons historiquement focalisé nos efforts sur des syndromes apparentés de malformations concernant la peau parmi d'autres systèmes d'organes. Ceux-ci sont le résultat de mutations post-zygotiques retrouvées également dans de multiples cancers à l'âge adulte. Cela veut dire que quelque chose dans le moment de la survenue de la mutation (avant ou après la naissance) ou, plus exactement, dans le contexte autour de et dans la cellule mutée à ces moments de sa vie, qui influe sur combien de telles mutations sont réellement "oncogéniques". Dans les deux cas de figure, l'organisme humain est en réalité un mosaïque de cellules atteintes par de telles mutations et d'autres qui ne le sont pas. Leurs interactions au fil du développement embryonnaire et foetal peuvent être pathogéniques.
Nos modèles de souris montrent qu'il est possible de phénocopier d'autres syndromes congénitaux pédiatriques en dirigeant les mêmes mutations "oncogéniques" à des dérivés distincts de la crête neurale multipotente au fil de sa différenciation. Ainsi, leurs dérivés dans la peau mais également dans le système nerveux périphérique, dans le coeur, dans l'hypophyse ou dans le crâne peuvent en montrer les effets. La caractérisation de ces modèles et la recherche de mutations équivalentes dans les tissus atteints dans des cohortes de patients pertinents pourront mener à un diagnostic amélioré et un traitement plus ciblé pour de nombreuses maladies rares. Nous complétons ces travaux par des études sur des cellules issues de patients et des cellules souches pluripotentes induites puis redifférenciées.
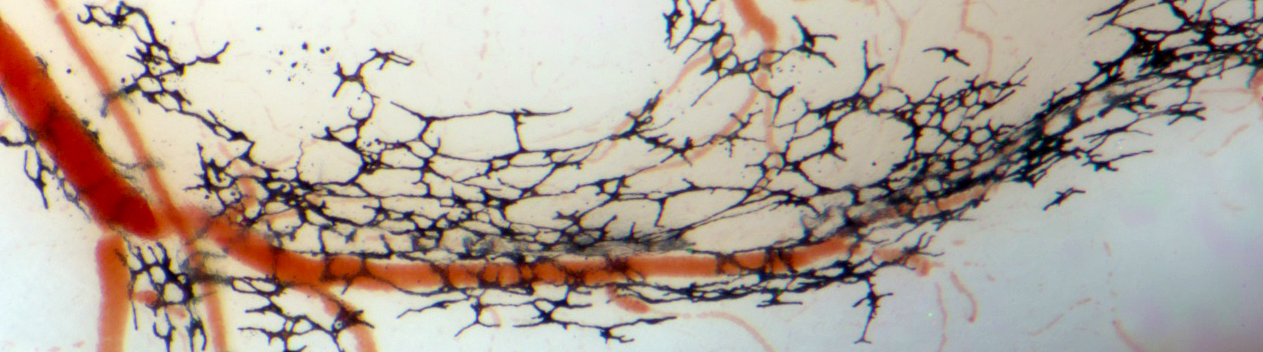
Le naevus géant (ou moins géant) congénital (NGC) est une malformation visible d'une partie de la peau, présente à la naissance. Il peut être isolé, stable et restreint, ou associé en syndrome avec d'autres symptômes cutanés, neurologiques ou oncologiques.
Depuis plus de 10 ans, nous étudions les effets des voies de signalisation assez universelles mais qui sont activées dans les précurseurs embryologiques aux cellules pigmentaires des NGC avec de multiples approches expérimentales et ressources :
Ce projet a reçu un soutien de l'Union Européenne sous forme du consortion MELCAYA et sera grandement renforcé par le recrutement d'un chercheur post-doctorant, Dr Daniel Aldea, qui a publié et continue à mener des recherches sur les premisses de prédisposition aux complications.
Nous continuons à étudier en rapport avec les projets sur le NGC, les origines moléculaires et embryologiques de certaines malformations vasculaires afin d'identifier des stratégies pour prévenir leur évolution postnatale. Comme dans le NGC, des mutations somatiques de gènes codant des enzymes de la voie RAS-RAF (MAPK) ou dans la voie PI3K, propagées dans certains composants périvasculaires, sont à l'origine de ces maladies rares et sont parfois létales. De telles mutations semblent exister en épistase à un héritage de fond génétique permissif.
Ces recherches ont des implications importantes pour comprendre la pathophysiologie de certains syndromes rares pédiatriques qui touchent aux systèmes cardiovasculaire, neuroendocrinien ou craniofacial. Des travaux publiés du Dr Elise Marechal ont caractérisé des modèles de souris qui permettent d'en décortiquer les mécanismes.
Ce projet de consortium national avait démarré juste avant la pandémie de COVID-19 en collaboration avec le groupe Zaffran dans l'unité MMG. Il s'agit de cartographier les signatures moleculaires de chaque type de cellule dans le corps humain au cours du développement afin de mieux comprendre leur physiologie et leurs interactions au cours de la vie prénatale. Nous avons introduit des techniques de pointe à notre plateforme GBiM et nous avons contribué des données d'une précision jamais vue sur la composition cellulaire du coeur au fil du troisième mois de grossesse, quand il change d'un ébauche à un organe de plus en plus fonctionnel. Ces données sont nos contributions à l'effort international Human Cell Atlas, soutenues par l'AFM Téléthon et l'INSERM.
E-mail:
Téléphone: 04 91 32 49 37
MCUPH
E-mail:
Téléphone: 0413429023
PH Dr.
E-mail:
TCH
E-mail:
Téléphone: 04 91 32 41 33
DOCTORANT(E)
E-mail:




