Vascular and pigmented neurocristopathies
Team Leader
H. ETCHEVERS
H. ETCHEVERS

We study how certain kinds of birth defects occur. Those affecting derivatives of the stem cell population known as the neural crest are called congenital neurocristopathies. The palate, eyes and/or heart are frequently affected, but dozens of rarer diseases present also symptoms in the skin, gut or the nervous system. While the genetic bases of many specific neurocristopathies over the last decade have been found, most remain without molecular diagnosis and are relatively poorly defined as clinical entities due to their diverse presentations and rarity.
We study a subset of malformation syndromes due to mutations also found in many adult cancers. Such mutations lead to constant activation of normally temperorarily active enzymes in only some cell types and can be lethal, depending on when they occur. The result in survivors is inappropriate growth factor signaling, leading to effects on cell identities and tissue growth. The resulting organism is a mosaic of affected (mutated) and unaffected (non-mutated) cells. Their interactions during development can lead to diseases that appear very different from, but are mechanistically related to and sometimes predispose to, certain cancers.
Mouse models used by our group phenocopy (reproduce many aspects of) syndromes found in human fetuses or children by directing the same types of mutations to distinct multipotent neural crest derivatives. This can affect not only their progeny in the skin but also in the heart, the peripheral nervous system, the pituitary or the skull. Characterizing these models and searching for equivalent mutations in relevant patient cohorts should lead to improved diagnoses and new therapeutic approaches for congenital neurocristopathies by repurposing drugs used in targeted chemotherapies.
Dr. Daniel Aldea, a senior postdoctoral scientist, has been carrying this project forward as part of the EU-funded MELCAYA consortium.
The large and giant congenital melanocytic nevus (CMN) is a visibly conspicuous malformation of the skin, present at birth. It can present as a restricted, stable and benign tumor, or be associated in syndromic form with additional cutaneous, neurological or oncological symptoms.
We study the effects of the molecular signaling pathways shown to be present in CMN in the embryological precursors to pigment cells using multiple systems:
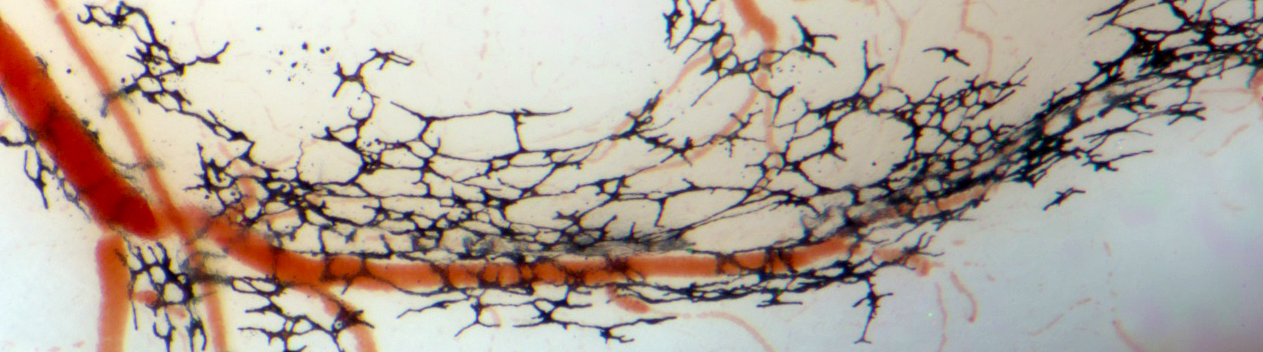
Somatic mutations in genes encoding one or more effectors of the RAS-RAF and PI3K signaling pathways, and propagated in the cellular components of blood vessels, cause rare, defiguring and sometimes lethal vascular malformations. Dr. Elise Marechal, during her Ph.D. work with us, helped delineate many of these phenotypes.
Our transgenic mouse and human cellular models for the expression of such mutations in neural crest lineages implicates these signaling pathways not only in vascular but also craniofacial malformations affecting the skull and palate, in progressive peripheral neuropathies (see 2025 preprint on bioRxiv, submitted for 2026 publication), and in neuroendocrine disorders.
This national consortium project launched just before the COVID-19 pandemic with the Zaffran group. It entailed mapping the specific transcriptomic signatures of each cell in the developing human body in order to better understand normal and disease-associated physiology during prenatal life. We introduced new technologies to the GBiM platform and contributed unprecedently detailed data on the cellular composition of the first-trimester heart as it turns from a primordium into a functional and vital organ. This data contributes to the international Human Cell Atlas effort and was published, with a cover illustration, in the journal Development in 2025.





Mail:
Telephone: 04 91 32 49 37
MCUPH
Mail:
Telephone: 0413429023
PH Dr.
Mail:
TCH
Mail:
Telephone: 04 91 32 41 33
DOCTORANT(E)
Mail: